Acute Disseminated Encephalomyelitis (ADEM): An Evolving Story in Neuroimmunology

TL;DR
ADEM remains one of the most important acquired demyelinating syndromes in children, yet our understanding of the disease is rapidly evolving. New discoveries in neuroimmunology are reshaping how clinicians think about diagnosis, recovery, and relapse risk, while raising important questions for patients and families navigating life after the acute illness. This post reviews the emerging science, current treatment landscape, and key opportunities for future research.
Crossing Paths with Rare Diseases
Until recently, I had never heard of acute disseminated encephalomyelitis (ADEM).
Like many families, we were introduced to it unexpectedly. My four-year-old nephew developed what initially appeared to be a routine childhood illness—fatigue, fever, and a brief fainting episode. Within hours, he was admitted to the intensive care unit. Days later, he was diagnosed with ADEM.
Three weeks and countless tests, scans, medications, and therapy sessions later, he was finally strong enough to go home.
Today, our family finds itself in a place that will be familiar to many affected by rare diseases: the acute crisis has passed, rehabilitation is underway, and life is gradually settling into a new normal. Yet many questions remain. What caused this? What does recovery really look like? What do we know about the chances of recurrence? And perhaps most importantly, what don’t we know?
My nephew, meanwhile, seems less concerned with these questions. If anything, he may miss the unlimited screen time, daily pancakes, and steady stream of visitors that came with his hospital stay.
For families, ADEM often begins as a frightening and bewildering diagnosis. For clinicians and researchers, it represents an intriguing and rapidly evolving area of neuroimmunology.
ADEM is rare enough that many clinicians encounter only a handful of cases during their careers, yet it remains one of the most common acquired demyelinating syndromes in children. For decades, ADEM was viewed primarily as an acute, post-infectious inflammatory disorder with a generally favorable prognosis. Today, that understanding is evolving.
Advances in neuroimmunology have transformed how researchers think about inflammatory diseases of the central nervous system (CNS). The discovery of disease-associated autoantibodies, improvements in neuroimaging, and growing recognition of long-term neurocognitive outcomes have revealed a more complex picture of ADEM than previously appreciated. In particular, the emergence of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) has challenged longstanding assumptions about disease classification, prognosis, and treatment.
For researchers and clinicians, ADEM offers a lens through which to study the intersection of infection, immune regulation, CNS repair, and precision medicine. For patients and families, however, the questions remain practical and deeply personal.
Disease Biology
At its core, ADEM is a disorder of immune-mediated inflammation affecting the brain and spinal cord. The condition is characterized by widespread demyelination, a process in which the immune system damages myelin, the insulating layer that surrounds nerve fibers and enables efficient communication throughout the nervous system.
Clinically, ADEM is defined by a combination of encephalopathy—confusion, irritability, somnolence, behavioral change, or coma—and polyfocal neurologic deficits, such as weakness, ataxia, optic neuritis, brainstem signs, seizures, sensory symptoms, or urinary retention. MRI typically shows large, bilateral, asymmetric T2/FLAIR hyperintense lesions involving subcortical and deep white matter, deep gray nuclei such as thalami and basal ganglia, brainstem, cerebellum, and sometimes spinal cord.
A key practical point is that ADEM is a diagnosis of exclusion in the acute setting. Viral encephalitis, bacterial CNS infection, multiple sclerosis, NMOSD, MOGAD, vasculitis, metabolic disease, and toxic leukoencephalopathies must be considered.
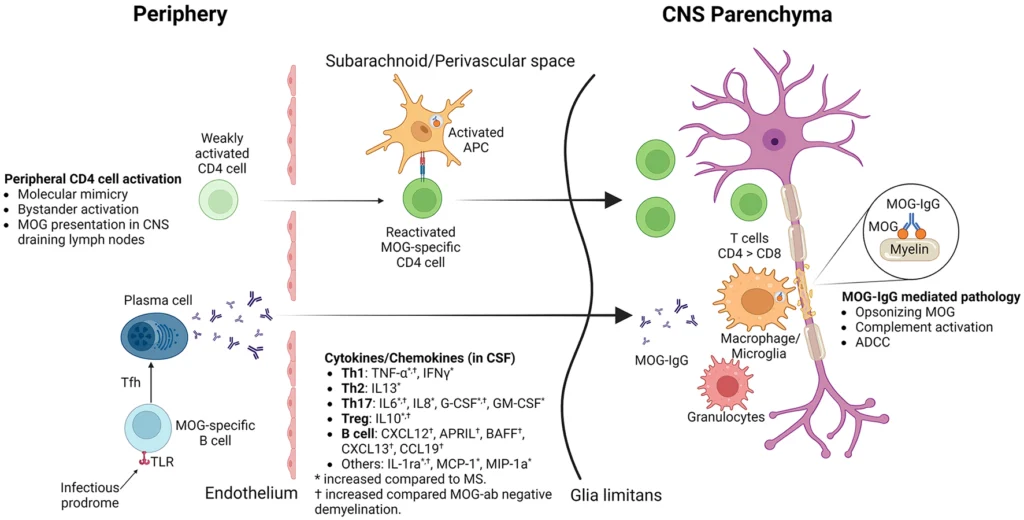
Historically, ADEM has been considered a post-infectious autoimmune syndrome. In many cases, neurological symptoms develop days to weeks after a viral or bacterial infection. This temporal relationship led to the hypothesis of molecular mimicry, in which immune responses directed against infectious agents inadvertently cross-react with components of CNS tissue (Pohl et al., 2016).
While this framework remains relevant, recent discoveries suggest the biology is more nuanced.
One of the most important developments in the field has been the identification of antibodies against myelin oligodendrocyte glycoprotein (MOG). Studies have shown that a substantial proportion of pediatric patients presenting with ADEM are positive for MOG antibodies, linking what was once considered a single clinical syndrome to a broader category of antibody-mediated neuroinflammatory disease (Brenton, 2024).
This distinction has important implications. It suggests that ADEM may not represent a single disease entity but rather a common clinical presentation arising from multiple immunological pathways. Some patients experience a monophasic inflammatory event with complete recovery, while others may develop recurrent disease consistent with MOGAD.
| Feature | Classic/MOG-negative ADEM | Mog-IgG-positive ADEM/MOGAD-ADEM |
|---|---|---|
| Typical age | Predominantly pediatric, but adults can be affected. | Particularly common in children; adult ADEM-like MOGAD occurs but is less common. |
| Trigger | Often post-infectious; vaccination-associated cases are less common today. | May also follow infection/vaccination, but antibody-mediated demyelination is central. |
| Clinical phenotype | Encephalopathy + multifocal deficits; often monophasic. | ADEM may be followed by optic neuritis, myelitis, cortical encephalitis, or multiphasic disease. |
| Relapse risk | Usually monophasic; relapse suggests multiphasic ADEM, MOGAD, MS, or NMOSD. | Relapse risk is higher than historically assumed for MOGAD; global review data suggest relapse proportion increases with longer follow-up. |
| Testing | MRI/CSF/infectious workup; MOG testing increasingly recommended. | Serum MOG-IgG cell-based assay is preferred; CSF-restricted MOG-IgG is uncommon and can be misleading. |
| Long-term management | Often no chronic immunotherapy after one monophasic event. | Maintenance therapy may be considered after relapse or high-risk features; IVIG, rituximab, mycophenolate, azathioprine, and tocilizumab are used off-label. |
This shift mirrors a broader trend across medicine. Diseases once defined solely by clinical symptoms are increasingly being reclassified according to their underlying biology. In neuroimmunology, ADEM appears to be following a similar trajectory.
Epidemiology
ADEM remains a rare disease, with reported incidence estimates ranging from approximately 0.07 to 0.64 cases per 100,000 children annually, depending on the population studied (Cole et al., 2019).
The disease is predominantly pediatric, with most cases occurring between 5 and 8 years of age. A slight male predominance has been observed, and incidence often peaks during winter and spring, reflecting seasonal patterns of infectious illnesses (Cole et al., 2019).
From an epidemiological perspective, rarity presents both challenges and opportunities. Small patient populations complicate clinical trial design, limit natural history data, and make longitudinal outcome studies difficult to conduct. At the same time, rare diseases often serve as powerful models for understanding fundamental biological mechanisms.
Importantly, epidemiological studies have also challenged the longstanding assumption that ADEM is uniformly benign.
In many respects, ADEM exemplifies this dynamic. Despite its low prevalence, insights gained from studying immune-mediated demyelination have influenced broader thinking about CNS inflammation, autoimmune disease, and neural repair.
Most patients experience substantial neurological recovery. However, growing evidence suggests that some children continue to experience cognitive, behavioral, educational, and psychosocial difficulties long after acute symptoms resolve (Koelman et al., 2020). These findings have expanded the definition of recovery beyond physical function alone and underscore the importance of long-term follow-up.
Current Treatment Landscape
Despite advances in our understanding of disease biology, treatment of ADEM remains remarkably unchanged.
High-dose intravenous corticosteroids continue to serve as the standard first-line intervention. Their objective is to suppress acute inflammation quickly enough to minimize tissue injury and preserve neurological function (Anilkumar et al., 2024).
For patients who fail to respond adequately, intravenous immunoglobulin (IVIG) and plasma exchange (PLEX) are commonly employed as second- and third-line therapies. These interventions have become standard components of clinical practice despite a relative lack of prospective randomized evidence.
| Treatment | Typical Role | Evidence/Clinical Use |
|---|---|---|
| Supportive care | Seizure control, ICU care if needed, rehab, bladder/bowel management, nutrition, prevention of complications. | Severe neurologic deficits, seizures, brainstem involvement, or altered consciousness may require ICU-level care. |
| Empiric antimicrobials/acyclovir | Used early when infectious encephalitis/meningitis remains possible. | Acute ADEM can resemble CNS infection, and treatment may begin before all infectious studies return. |
| High-dose IV methylprednisolone | First-line disease-directed therapy. | Standard regimen is methylprednisolone 30 mg/kg/day IV, maximum 1 g/day, for 3–5 days, followed by oral prednisone taper over 4–6 weeks. |
| Oral steroid taper | Prevents rebound inflammation/early relapse. | Early steroid discontinuation may increase relapse risk. |
| IVIG | Second-line for steroid-incomplete response or when steroids are contraindicated. | Dose commonly 2 g/kg over 2–5 days. |
| Plasma exchange | Severe, steroid/IVIG-refractory, fulminant, or hemorrhagic cases. | Usually 3–7 exchanges; often considered earlier in severe MOGAD/NMOSD-like attacks. |
| Cyclophosphamide | Rare refractory cases. | Used when severe inflammatory disease fails standard rescue therapy. |
| Rehabilitation and neuropsychology | Motor, cognitive, speech, vision, and school/work reintegration. | Cognitive impairment can persist after pediatric ADEM, even when motor recovery is good. |
This reality reflects a familiar challenge in rare disease medicine. Clinical urgency often outpaces the ability to generate definitive trial data.
From a therapeutic development perspective, ADEM remains largely dependent on broad immunosuppression rather than mechanism-specific intervention. The current approach treats inflammation effectively but does little to address underlying biological heterogeneity.
As biomarker discovery advances, this paradigm may begin to shift. The growing recognition of MOG-associated disease, coupled with increasing sophistication in immune profiling, raises the possibility that future treatment strategies could become more targeted and individualized.
The evolution of therapy in multiple sclerosis from nonspecific immunosuppression to highly selective immune modulation offers a useful precedent. Whether a similar transformation awaits ADEM remains an open question.
Clinical Trials
If disease biology is evolving rapidly, clinical evidence generation has progressed more slowly.
ADEM presents a challenging environment for traditional drug development. The disease is rare, frequently monophasic, and biologically heterogeneous. These characteristics complicate patient recruitment, endpoint selection, and trial execution.
This does not reflect a lack of scientific interest. Rather, it highlights the practical realities of studying uncommon pediatric neurological disorders.
ADEM-specific randomized trials remain limited because the syndrome is rare, acute, heterogeneous, and often monophasic. The active clinical development landscape is therefore centered on MOGAD, which includes ADEM phenotypes.
| Trial/Agent | Mechanism | Population | Status/Design | Why it Matters |
|---|---|---|---|---|
| Rozanolixizumab – cosMOG, NCT05063162 | FcRn inhibitor that lowers circulating IgG, including pathogenic autoantibodies. | Adults 18–89 with relapsing MOGAD and documented serum MOG antibody positivity. | Phase 3, randomized, double-blind, placebo-controlled, multicenter trial with open-label extension; active, not recruiting; actual enrollment 113; estimated primary completion May 2027. | First pivotal-style trial directly testing whether IgG reduction can prevent MOGAD relapse. |
| Tocilizumab – TOMATO, NCT06452537 | IL-6 receptor blockade. | Patients ≥12 years with seropositive MOGAD and recent relapse history. | Randomized, controlled, multicenter Phase 2/3 study; active, not recruiting; planned enrollment 102; compares tocilizumab plus prednisone vs prednisone; primary outcome is time to first MOGAD relapse up to 60 weeks. | Tests a rational cytokine-targeted approach; IL-6 is implicated in B-cell activation, blood–brain barrier dysfunction, leukocyte migration, and autoantibody production 14. |
| Satralizumab – NCT05271409 | IL-6 receptor blockade, long-acting monoclonal antibody. | MOGAD population. | Study objective is to evaluate satralizumab vs placebo based on time to first protocol-defined relapse. | Builds on IL-6 pathway success in NMOSD and observational interest in MOGAD. |
| Tocilizumab observational/open-label evidence | IL-6 receptor blockade. | Refractory MOGAD, including MOG optic neuritis and highly relapsing disease. | A review of 24 studies found reduced annualized relapse rates and improved/stabilized neurologic outcomes in many reports, but evidence remains small and observational. | Supports IL-6 blockade as a leading near-term pathway, especially in steroid-dependent or refractory disease. |
| Autologous HSCT case-level evidence | Immune reset. | Highly aggressive, treatment-refractory MOGAD. | Case-level evidence only; not a routine pathway. | Illustrates need for escalation pathways in rare, severe MOGAD. |
By identifying biologically defined patient populations, researchers are beginning to move beyond syndromic classifications toward more precise disease categories. This transition has the potential to improve patient stratification, facilitate biomarker-driven studies, and create more homogeneous cohorts for therapeutic investigation.
Several research priorities have emerged across the field:
- Validation of prognostic biomarkers
- Better characterization of MOG-positive versus MOG-negative disease
- Long-term neurocognitive outcome assessment
- Development of standardized clinical endpoints
- Identification of predictors of relapse and disability
- Evaluation of targeted immunomodulatory therapies
Notably, many of these priorities extend beyond traditional neurological measures.
Increasingly, investigators are asking questions about school performance, cognitive development, quality of life, mental health, and functional independence. These outcomes may ultimately prove as important to patients and families as MRI findings or relapse rates.
Opportunities and White Space
For a disease that has been recognized for decades, ADEM remains surprisingly underexplored.
- One of the largest opportunities lies in biomarker development. Although MRI and MOG antibody testing have improved diagnostic confidence, clinicians still lack robust tools for predicting disease course, treatment response, or long-term outcomes. The ability to identify high-risk patients early could fundamentally change management strategies.
- A second opportunity involves disease stratification. Increasing evidence suggests that ADEM represents a clinical syndrome rather than a single biological entity. As our understanding of immune pathways expands, future classification systems may distinguish patient subgroups based on molecular and immunological characteristics rather than clinical presentation alone.
- Third, there is a growing need for longitudinal outcomes research. Historically, studies have focused on acute recovery. Yet families often describe challenges that emerge months or years later, including fatigue, attention deficits, executive dysfunction, learning difficulties, and emotional health concerns. These dimensions remain underrepresented in both clinical trials and routine follow-up care.
- Finally, ADEM highlights a broader opportunity within neuroimmunology: integrating biological discovery with patient-centered outcomes. The field has become increasingly sophisticated in measuring antibodies, cytokines, and imaging biomarkers. Equally important is understanding how these biological findings translate into meaningful improvements in everyday life.
Looking Ahead
ADEM sits at an interesting crossroads in modern medicine.
It remains a rare disease, but one that continues to generate outsized scientific interest. The condition touches many of the themes shaping contemporary neuroimmunology: immune dysregulation, biomarker discovery, disease heterogeneity, precision medicine, and long-term neurological recovery.
Perhaps most importantly, ADEM reminds us that disease classification is rarely static. As new biological insights emerge, familiar diagnoses often reveal unexpected complexity.
The story of ADEM is increasingly becoming a story about understanding that complexity.
For researchers, it represents an opportunity to deepen our understanding of CNS autoimmunity. For clinicians, it highlights the importance of looking beyond acute recovery. For industry and clinical development teams, it illustrates the challenges—and potential rewards—of advancing therapies in rare neuroinflammatory diseases.
And for patients and families, continued progress offers something even more important: the prospect of clearer answers, more precise treatments, and a better understanding of what recovery truly means.
Thanks for reading, I’d love to stay connected.
© Copyright 2026. Powered by Bluehost.